Sebagai Sindrom Bourneville-Pringle segitiga tumor otak dengan epilepsi dan kelewatan perkembangan, lesi kulit dan pertumbuhan pada sistem organ lain diketahui. Penyakit ini disebabkan oleh mutasi dua gen TSC1 dan TSC2. Terapi adalah simtomatik dengan fokus pada epilepsi.
Apa itu Sindrom Bourneville-Pringle?

© Henrie - stock.adobe.com
Istilah perubatan Bourneville-Pringle syndrome adalah sinonim untuk sklerosis tuberous. Fenomena patologi ini termasuk dalam kumpulan penyakit keturunan dan dicirikan oleh tumor jinak pada wajah, otak dan sistem organ, oleh ketidakupayaan intelektual dan serangan epilepsi.
Penyebaran sklerosis tuberous pada bayi baru lahir adalah sekitar satu kes pada 8.000 bayi. Ahli neurologi Perancis Désiré-Magloire Bourneville dan Édouard Brissaud pertama kali menggambarkan penyakit ini bersama dengan pakar dermatologi Britain, John James Pringle pada abad ke-19. Nama Bourneville-Pringle Syndrome telah wujud demi mereka.
Di dunia berbahasa Inggeris, kompleks gejala disebut Kompleks Sklerosis Tuberous. Secara klinikal, kompleks ini dicirikan oleh triad simptomatik dengan gejala yang disebutkan di atas. Bentuk khas sindrom adalah sindrom gen bersebelahan.
sebab-sebab
Kluster keluarga telah diperhatikan berkaitan dengan sindrom Bourneville-Pringle, nampaknya berdasarkan warisan dominan autosomal. Walau bagaimanapun, pada separuh daripada semua kes, penyakit ini berdasarkan mutasi genetik baru sebagai penyebabnya. Oleh itu, kadar mutasi spontan sekurang-kurangnya setinggi mutasi yang diwarisi.
Dalam kes keluarga, mutasi pada gen TSC1 pada lokus Chr.9q34 dan pada gen TSC2 pada lokus Chr.16p13 diperhatikan dengan frekuensi yang sama. Kejadian sporadis hampir secara eksklusif terhad kepada mutasi baru dalam gen TSC2. Kedua-dua gen adalah gen penekan tumor dan oleh itu terlibat dalam menekan pertumbuhan sel. Produk gen mereka adalah hamartin dan tuberin, fungsinya belum dapat dijelaskan secara meyakinkan.
Mutasi dalam konteks sindrom Bourneville-Pringle disebarkan ke seluruh gen ekson yang disebutkan dan boleh sesuai dengan jenis mutasi apa pun. Hanya penghapusan besar dalam gen TSC2 pada satu atau lebih ekson yang belum diperhatikan. Bentuk khas sindrom gen bersebelahan mempengaruhi gen TSC2 dan gen PKD1.
Gejala, penyakit & tanda
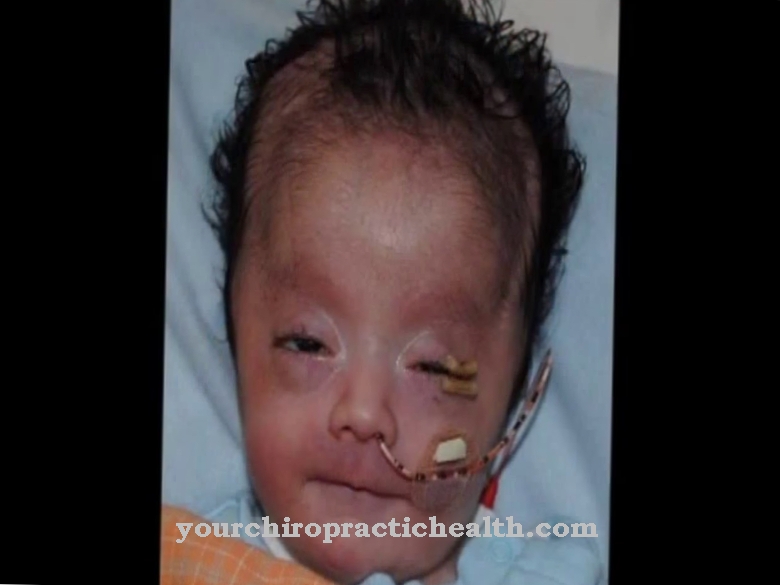
Sklerosis tuberus dicirikan oleh beberapa bidang pembezaan tisu yang tidak normal, yang disebut hamartiae, yang bervariasi di lokasi berkenaan dengan sistem organ. Kriteria utama penyakit ini merangkumi angiofibromas muka dan nevi tisu penghubung di kawasan dahi, angiofibromas tidak traumatik, sekurang-kurangnya tiga bintik hipomelanotik, nevi tisu penghubung sakrum dan beberapa hamartoma pada retina.
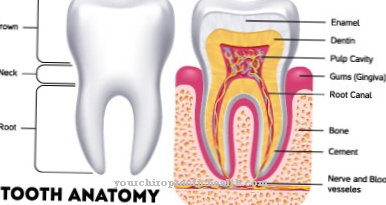
Selain displasia kortikal, terdapat juga nodul subependymal, gejala sel gergasi subependymal dan rhabdomyoma jantung. Sebagai tambahan, limfangiomyomatosis paru-paru dan angiomyolipoma buah pinggang boleh dinamakan sebagai kriteria utama. Mengiringi simptom, pesakit biasanya mengalami kecacatan enamel gigi, polip rektum atau pembentukan kista osseus.
Di samping itu, simptom-simptom mungkin disertai dengan pengecutan perkara otak yang tidak biasa. Perkara yang sama berlaku untuk fibroid gingiva, depigmentasi dan sista buah pinggang. Triad sindrom terbahagi kepada perubahan kulit simptomatik, kerosakan otak dengan gangguan perkembangan dan epilepsi dan gejala sistem organ lain.
Diagnosis & kursus
Untuk mendiagnosis sklerosis tuberous, doktor menunjukkan kepada pesakit sama ada dua kriteria utama penyakit ini atau satu gejala utama dengan dua kriteria sekunder. Perubahan pada otak biasanya dikesan paling awal dan biasanya ditunjukkan menggunakan pencitraan seperti MRI. Analisis genetik molekul dapat mengesahkan diagnosis sindrom yang disyaki dan mengesampingkan sindrom serupa dari diagnosis pembezaan.
Prognosisnya baik untuk pesakit dengan sindrom Bourneville-Pringle yang agak ringan. Kebanyakan pesakit dengan BPD ringan menjalani kehidupan normal. Orang yang terjejas oleh BPD yang teruk dan dengan itu epilepsi yang teruk, gangguan kognitif yang melampau dan sebilangan besar tumor mempunyai prognosis yang lebih buruk dan mungkin mengharapkan kesan pemendekan hidup.
Komplikasi
Dalam sindrom Bourneville-Pringle atau sklerosis tuberous, sistem organ yang berbeza terjejas dan boleh mempunyai komplikasi yang berbeza. Di satu pihak, penyakit ini terutama mempengaruhi sistem saraf pusat dan otak. Mereka yang terkena menderita epilepsi, terutama pada masa kanak-kanak. Kejang separa adalah yang paling biasa, tetapi juga boleh digeneralisasikan.
Sekiranya tidak dirawat, epilepsi kanak-kanak boleh berkembang menjadi sindrom Lennox-Gausaut. Orang yang berkenaan menderita sawan dan ketidakhadiran tonik beberapa kali sehari, yang dalam keadaan terburuk boleh berubah menjadi epileptikus status, kecemasan perubatan. Kadang-kadang gangguan perkembangan mental juga dapat diperhatikan pada anak.
Selanjutnya, seorang pesakit boleh mengalami peningkatan tekanan intrakranial dalam perjalanan penyakit ini. Ini membawa kepada sakit kepala yang teruk dan kesedaran yang lemah. Dalam kes yang paling teruk, pusat kawalan penting boleh terperangkap di kawasan medula memanjang (medulla oblongata), yang boleh menyebabkan kegagalan pernafasan.
Sklerosis tuberus juga boleh menjadi penyebab kista ginjal atau tumor ganas, yang boleh menyebabkan kegagalan buah pinggang (kekurangan buah pinggang). Ini sangat mengehadkan kualiti hidup dan pesakit mungkin perlu menjalani dialisis atau transplantasi. Rhabdomyoma intracardiac dapat berkembang di jantung, yang boleh menyebabkan aritmia jantung atau bahkan kematian jantung.
Bilakah anda harus berjumpa doktor?
Sekiranya kejang epilepsi dan gangguan kognitif berterusan, doktor harus berunding. Dia dapat menggunakan pemeriksaan ultrasound untuk menentukan apakah sindrom Bourneville-Pringle adalah penyebabnya. Diagnosis penyakit tumor yang disasarkan hanya mungkin dilakukan setelah anamnesis menyeluruh. Epilepsi ciri sudah dapat ditentukan pada bulan-bulan pertama kehidupan.
Pakar pediatrik kemudian akan melakukan pemeriksaan rutin dan dengan cepat mendiagnosis sindrom Bourneville-Pringle. Sekiranya kejang epilepsi tidak berlaku, diagnosisnya lebih sukar. Sebarang gangguan perkembangan dan masalah tingkah laku sering hanya berlaku pada masa kanak-kanak atau remaja. Pada dasarnya: jika kanak-kanak berkelakuan tidak normal, mengalami kesukaran untuk belajar atau menunjukkan kelemahan lain, pakar pediatrik mesti mendapatkan nasihat.
Tanda-tanda amaran lain yang memerlukan penjelasan perubatan adalah peningkatan perubahan kulit seperti poplars kemerahan atau bintik-bintik berbentuk daun pada kulit. Selanjutnya, tumor kulit, benjolan dan kelainan lain dapat muncul. Sekiranya salah satu atau kedua ibu bapa menghidap sindrom Bourneville-Pringle, disarankan untuk mendapatkan penilaian perubatan semasa kehamilan.
Doktor & ahli terapi di kawasan anda
Rawatan & Terapi
Sejauh ini, sindrom Bourneville-Pringle tidak dapat diobati secara kausal, kerana hanya pendekatan terapi gen yang dapat dianggap sebagai terapi kausal dan pendekatan ini saat ini menjadi subjek kajian, tetapi belum disetujui untuk digunakan. Atas sebab ini, hanya terapi simptomatik yang tersedia untuk rawatan.
Terapi epilepsi adalah fokus terapi, kerana gejala inilah yang sangat merosakkan kualiti hidup mereka yang terjejas dan, dalam keadaan terburuk, menyebabkan kemerosotan keadaan kesihatan hingga mati. Rawatan epilepsi boleh diubati atau, dalam kes yang teruk, pembedahan sejauh mungkin.
Sebagai contoh, pemisahan kedua belahan otak melalui pembedahan membuang korpus callosum telah menunjukkan kejayaan dalam terapi epilepsi pada masa lalu. Untuk bentuk yang lebih ringan, pemberian anti-epilepsi sering mencukupi. Sebagai tambahan kepada langkah-langkah rawatan ini, tumor mesti dikeluarkan dari sistem organ. Kerana ini kebanyakannya tumor jinak, penyinaran seterusnya biasanya tidak ditunjukkan.
Dalam kes tumor yang banyak, pemantauan rapi ditunjukkan untuk mengenal pasti kemungkinan perubahan yang membawa kepada malignan pada waktunya. Oleh kerana mereka yang terkena penyakit sering mengalami keterlambatan mental, langkah-langkah seperti intervensi awal juga merupakan langkah terapi yang sesuai. Perkembangan pertuturan dapat disokong dengan terapi pertuturan.
Kelewatan pengembangan motor dapat diatasi dengan langkah fisioterapi dan terapi pekerjaan. Sekiranya penyakit itu mengakibatkan tekanan psikologi bagi pesakit, psikoterapi juga berguna.
Prospek & ramalan
Tidak ada terapi kuratif untuk sindrom Bourneville-Pringle. Hanya rawatan simptomatik yang mungkin. Keterukan penyakit ini berbeza bagi setiap pesakit. Sebagai peraturan, jangka hayat adalah normal. Walau bagaimanapun, ini dapat dikurangkan dengan serangan epilepsi yang kerap, keterbelakangan mental yang teruk dan degenerasi malignan dari tumor yang ada.
Terapi ini terhad kepada rawatan sawan epilepsi. Sebagai sebahagian daripada penyakit ini, semua jenis kejang berlaku pada epilepsi. Perkaitan antara perkembangan kognitif dan kekerapan kejang telah diperhatikan. Orang dewasa terutamanya mengalami kejang fokus sekunder umum.
Secara keseluruhan, terdapat gangguan perkembangan yang menampakkan diri dalam gangguan bahasa, pergerakan dan pembelajaran. Hasil kecerdasan individu yang terjejas dapat berkembang secara berbeza. Walaupun ini adalah normal pada separuh pesakit, sekitar 31 peratus pesakit mencapai maksimum 21 orang.
Perubahan kulit juga berbeza dan bergantung pada usia. Ini adalah adenoma sebum. Rawatan kosmetik adenoma dilakukan dengan penyingkiran pembedahan atau penyinaran laser. Selalunya terdapat juga angiomyolipoma, tumor jinak pada tisu ginjal. Tumor jinak juga boleh berkembang di otot jantung yang berlarutan. Organ lain seperti paru-paru juga boleh terkena tumor. Kemerosotan malignan sangat jarang berlaku.
pencegahan
Sejauh ini, sindrom Bourneville-Pringle hanya dapat dicegah jika, dalam perancangan keluarga, pasangan dapat menggunakan ujian genetik molekul untuk menilai risiko mereka terhadap anak yang sakit dan, jika ada peningkatan risiko, memutuskan untuk tidak memiliki anak sendiri.
Anda boleh melakukannya sendiri
Sklerosis tuberous, juga dikenal sebagai sindrom Bourneville-Pringle, adalah penyakit genetik yang belum dapat diobati secara kausal. Oleh itu, langkah-langkah terapi bermula dengan gejala.
Epilepsi adalah salah satu kesan sampingan yang paling menyusahkan yang biasanya menjejaskan kualiti hidup mereka yang terjejas. Sebagai tambahan kepada rawatan ubat dengan ubat anti-epilepsi, mereka yang terjejas sering menyumbang kepada fakta bahawa kejang berlaku lebih jarang atau kurang teruk melalui gaya hidup mereka. Pesakit harus menyimpan buku harian epilepsi untuk mengetahui sama ada faktor dalam kehidupan seharian mereka yang menyebabkan kejang.
Faktor-faktor seperti itu boleh berbeza sama sekali. Makanan tertentu, alkohol, ubat-ubatan yang mengubah fikiran serta kurang tidur, tekanan, perasaan cemas yang teruk atau, pada wanita, tempoh haid. Faktor kritikal harus dielakkan sejauh mungkin. Banyak penderita dan saudara-mara mereka juga mendapat manfaat daripada menyertai kumpulan bantuan diri untuk epilepsi, yang kini wujud di banyak bandar di Jerman.
Selalunya, mereka yang terkena sklerosis tuberous juga mengalami perkembangan intelektual yang terbelakang. Akibat negatifnya dapat diatasi dengan intervensi awal yang mencukupi. Ibu bapa boleh mendapatkan nasihat daripada pakar pediatrik atau pejabat kebajikan remaja. Sekiranya perkembangan kemahiran motorik juga terganggu, langkah-langkah pekerjaan dan fisioterapeutik akan membantu. Sekiranya perkembangan bahasa yang tertangguh, pakar terapi pertuturan harus berunding.


.jpg)



.jpg)

.jpg)

.jpg)



.jpg)