The Sindrom Muenke dicirikan oleh kraniosyostosis jahitan koronari, yang disebabkan oleh mutasi pada gen FGFR3. Kelainan ini diwarisi sebagai sifat autosomal dominan dan sering disertai oleh ekstremitas yang tidak normal. Rawatan biasanya sesuai dengan prosedur pembedahan.
Apa itu Sindrom Muenke?

© flashmovie - stock.adobe.com
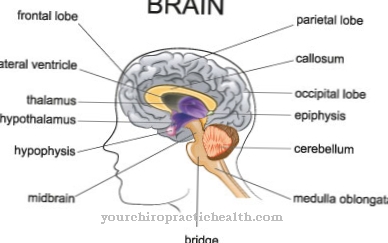
Dalam kraniosynostosis, satu atau lebih jahitan kranial muncul sebelum waktunya semasa perkembangan embrio dan dengan itu menghalang pertumbuhan fisiologi tengkorak dan otak. Banyak penyakit dari kumpulan sindrom kecacatan kongenital dengan penglibatan wajah yang paling banyak mengandungi kraniosyostosis seperti itu. Penyakit seperti itu Sindrom Sindrom Jahitan Koronari, juga dipanggil Sindrom Muenke dikenali.
Penyakit ini pertama kali dijelaskan pada tahun 1997. M. Muenke dan rakan-rakannya adalah yang pertama menerangkannya. Sindrom Muenke dicirikan oleh kraniosynostosis jahitan koronari dan juga merangkumi perubahan rangka pada tulang tarsus dan karpal. Penyebaran sindrom pada masa ini tidak diketahui. Manifestasi berlaku pada masa neonatal awal atau paling lambat pada usia awal kanak-kanak. Walaupun penyakit ini belum diteliti secara konklusif, penyebabnya kini telah dijelaskan.
sebab-sebab
Dalam banyak kes, sindrom Muenke boleh dikaitkan dengan pengumpulan keluarga. Dalam kes ini, pewarisan menyerupai warisan dominan autosomal. Namun, kes-kes juga telah didokumentasikan di mana sindrom tersebut nampaknya berlaku secara sporadis. Penyebabnya adalah mutasi genetik, yang dalam kes sporadis mungkin sesuai dengan mutasi baru. Lokasi mutasi juga dianggap dikenal pasti.
Penyakit ini dikatakan berdasarkan mutasi pada gen FGFR3, yang dapat dijumpai di lokus gen 4p16.3. Sindrom lain juga dikaitkan dengan gen FGFR3. Salah satu contohnya ialah sindrom Apert yang disebut. Gen memberi kod untuk reseptor faktor pertumbuhan fibroblas 3 dalam DNA. Sedikit yang diketahui mengenai kesan fisiologi faktor pertumbuhan FGF-3.
Menurut spekulasi, FGF-3 adalah faktor penentu terutama untuk tempoh embrio. Mutasi reseptor mungkin bermaksud bahawa faktor pertumbuhan tidak mengikat secukupnya semasa perkembangan embrio.
Gejala, penyakit & tanda
Pesakit sindrom Muenke mengalami pelbagai gejala. Oleh kerana penutupan jahitan koronari pada tahap awal, mereka yang terkena mempunyai bentuk kepala yang tidak normal, yang juga terlihat pada anomali wajah. Selain diameter tengkorak anterio-posterior yang dipendekkan, soket mata biasanya kurang dalam.
Gejala ini sering dikaitkan dengan hipoplasia rahang atas. Sekiranya jahitan koronari ditutup di satu sisi, soket mata diratakan pada sisi yang sesuai. Biasanya kecerdasan pesakit tidak terjejas oleh sindrom. Fusi tulang tangan atau tulang tarsal terdapat pada anggota badan. Malregasi juga dapat dibayangkan pada tulang karpal.
Epifisis kerucut juga merupakan salah satu gejala yang mungkin berlaku. Dalam beberapa kes, gambaran klinikal pesakit juga dikaitkan dengan osteochondromas. Fenotipik dan pertindihan simptomatik dengan sindrom lain seperti sindrom Pfeiffer, sindrom Jackson-Weiss atau sindrom Saethre-Chotzen adalah manifestasi yang dapat difahami secara klinikal.
Diagnosis & perjalanan penyakit
Diagnosis sindrom Muenke biasanya dibuat pada masa neonatal, kerana penyakit ini dapat dikenali lebih awal dengan diagnosis visual. Kira-kira satu pesakit dalam 15,000 bayi baru lahir menderita sinostosis jahitan koronari. Walau bagaimanapun, fenomena ini tidak semestinya disebabkan oleh sindrom Muenke.
Oleh itu, diagnostik memerlukan pengesanan mutasi patogen pada gen FGFR3. Tangan dan kaki pesakit mungkin kelihatan normal secara radiologis, jadi tidak mencukupi untuk mencari kelainan dalam hal diagnosis. Pada prinsipnya, semua anak dengan sinostosis koronari dapat disaring untuk mutasi P250R tertentu.
Pemeriksaan ini sesuai dengan analisis genetik molekul. Pengecualian mutasi tidak semestinya pesakit tidak mengalami sindrom Muenke. Dalam beberapa kes, mutasi tidak dapat dikesan pada mereka yang terjejas. Walau bagaimanapun, buktinya dianggap dapat memastikan diagnosis. Prognosis untuk pesakit wanita kurang digemari.
Komplikasi
Akibat sindrom Muenke, mereka yang terkena menderita pelbagai kecacatan dan kecacatan yang berlaku terutamanya di kepala dan wajah pesakit. Kecacatan ini sering menyebabkan keluhan psikologi dan kemurungan. Mereka yang terjejas sering mengalami kompleks rendah diri dan penurunan harga diri.
Gejala sering menimbulkan perasaan malu dan kanak-kanak khususnya boleh dipengaruhi oleh buli dan mengusik akibat sindrom Muenke. Namun, kecerdasan tidak terganggu, sehingga perkembangan mental pesakit berjalan tanpa komplikasi. Terdapat juga kecacatan anggota badan, sehingga terdapat sekatan dalam pelbagai aktiviti atau sekatan pergerakan dalam kehidupan seharian.
Kualiti hidup dikurangkan dengan ketara oleh sindrom Muenke. Tidak ada komplikasi lain dalam merawat sindrom Muenke. Sebagai peraturan, rawatan kausal tidak mungkin dilakukan, tetapi beberapa campur tangan pembedahan mesti dilakukan untuk mengelakkan kerosakan akibat selanjutnya.
Jangka hayat orang yang berkenaan biasanya tidak terhad. Rawatan juga boleh dilakukan sejurus selepas kelahiran. Dalam beberapa kes, ibu bapa kanak-kanak juga dipengaruhi oleh keluhan psikologi dari sindrom Muenke.
Bilakah anda harus berjumpa doktor?
Kecacatan ciri di kepala dan wajah adalah petunjuk yang jelas mengenai sindrom Muenke dan biasanya membawa kepada diagnosis sejurus selepas kelahiran. Sekiranya simptomnya ringan, doktor yang bertanggungjawab harus dimaklumkan mengenai sebarang gejala. Kadang-kadang, tulang karpal atau tarsal dapat tumbuh bersama, mengakibatkan gaya khas penyakit ini. Orang yang sudah mempunyai sejarah keluarga penyakit ini harus menjalani ujian genetik pada peringkat awal. Langkah-langkah rawatan yang diperlukan kemudian boleh dimulakan sejurus selepas kelahiran.
Ibu bapa kanak-kanak yang terjejas juga harus memberitahu doktor keluarga mereka mengenai gejala baru dan tingkah laku menyimpang kanak-kanak yang lain. Di samping itu, pemeriksaan rapi oleh pakar selalu diperlukan. Rawatan biasanya dilakukan di hospital atau di klinik pakar untuk penyakit genetik. Sebagai tambahan kepada doktor am, ortopedis atau internis boleh dipanggil. Untuk aduan kronik, terapi dan fisioterapi juga merupakan sebahagian daripada rawatan.
Terapi & Rawatan
Rawatan kausal tidak tersedia untuk pesakit dengan sindrom Muenke. Pendekatan terapi gen menawarkan harapan untuk terapi kausal, tetapi mereka belum mencapai tahap klinikal. Rawatannya hanya bersifat simtomatik dan dengan itu bergantung kepada gejala dalam kes individu. Hanya pilihan rawatan pembedahan yang boleh digunakan untuk memperbaiki keabnormalan tengkorak.
Pembedahan ini dirancang untuk membantu menghilangkan tekanan pada otak yang disebabkan oleh penutupan awal jahitan koronari. Saraf kranial lega dengan cara ini dan idealnya kurang atau tidak sama sekali dimampatkan selepas prosedur. Pilihan rawatan konservatif tersedia untuk kraniosynostosis yang kurang teruk. Contohnya, kanak-kanak yang lebih mudah terkena boleh diberi bentuk tengkorak yang harus mereka pakai untuk waktu yang lama.
Bentuk tengkorak ini cuba memodelkan tengkorak dengan secukupnya. Oleh kerana sindrom Muenke biasanya didiagnosis sejak bayi baru lahir, pembentukan semula konservatif seperti ini sangat berguna: bentuk kepala bayi masih dapat disesuaikan. Pemodelan konservatif akhirnya mempunyai matlamat yang sama dengan pemodelan operasi.
Pertumbuhan otak fisiologi harus diaktifkan dengan rawatan. Di samping itu, penampilan kepala disesuaikan dengan rata-rata. Gejala bersamaan seperti malformasi hujung kaki dapat dirawat secara pembedahan. Sekiranya mereka tidak menyekat atau menghalang orang yang terjejas, rawatan sedemikian tidak semestinya diperlukan.
Prospek & ramalan
Sindrom Muenke adalah penyakit jarang yang kini dapat dirawat secara pembedahan. Sekiranya penutupan awal jahitan koronari dikesan pada waktunya, prognosisnya baik. Segala kepincangan boleh dirawat secara pembedahan. Ubat yang sesuai boleh diresepkan untuk gejala seperti sakit atau gangguan kepekaan. Sindrom sinostosis jahitan koronari tidak mengurangkan jangka hayat.
Kualiti hidup dapat sedikit dibatasi oleh keluhan yang disebutkan serta kemungkinan parut di wajah dan kepala. Secara umum, prognosisnya positif. Kanak-kanak dengan sindrom Muenke sangat terdedah kepada gangguan pendengaran telinga dalam. Pendengaran yang lemah boleh menyebabkan masalah di kemudian hari, misalnya, ketika pesakit tidak lagi dapat memahami arahan atau mencari jalan dalam kehidupan seharian.
Kerana pengecualian dan penurunan harga diri, sebilangan orang sakit mengalami keluhan psikologi. Kesejahteraan dapat dikurangkan jika orang sakit tidak mempunyai jaringan sokongan yang baik dalam bentuk ibu bapa, saudara, rakan dan ahli terapi. Kanak-kanak yang menderita sindrom Muenke pasti memerlukan sokongan dalam kehidupan seharian agar dapat kekal sihat dari segi mental dan fizikal walaupun mengalami penyakit.
pencegahan
Sindrom Muenke tidak dapat dicegah kerana dipengaruhi oleh faktor genetik daripada faktor luaran. Kaunseling genetik semasa kehamilan adalah satu-satunya langkah pencegahan.
Penjagaan Selepas
Dalam kebanyakan kes, mereka yang terkena sindrom Muenke mempunyai langkah-langkah yang sangat sedikit dan hanya sangat terhad untuk rawatan susulan langsung. Pertama sekali, diagnosis awal dan cepat mengenai penyakit ini harus dibuat agar tidak ada komplikasi lebih lanjut. Oleh kerana sindrom Muenke adalah penyakit genetik, ia tidak dapat disembuhkan sepenuhnya.
Walau bagaimanapun, jika anda ingin mempunyai anak, ujian dan kaunseling genetik boleh berguna untuk mencegah sindrom itu berulang. Sebilangan besar mereka yang terjejas bergantung pada operasi. Pesakit mesti berehat dan beristirahat setelah operasi sedemikian, sebaiknya rehat di tempat tidur harus dijaga. Meletihkan atau melakukan aktiviti fizikal juga harus dielakkan agar tidak memberi tekanan kepada badan secara tidak perlu.
Dalam banyak kes, mereka yang terkena sindrom juga bergantung pada bantuan dan sokongan keluarga mereka sendiri dalam kehidupan seharian. Perbualan yang mesra dan intensif dengan keluarga dan rakan-rakan juga memberi kesan yang sangat positif terhadap perjalanan penyakit ini. Hal ini tidak biasa untuk mencegah perkembangan kemurungan atau gangguan psikologi yang lain.
Anda boleh melakukannya sendiri
Sindrom Muenke biasanya dirawat secara pembedahan dan dengan ubat-ubatan. Langkah pertolongan diri yang paling penting adalah menjaga badan selepas prosedur dan terus berunding dengan doktor yang bertanggungjawab. Ibu bapa kanak-kanak yang terjejas harus memerhatikan anak dengan berhati-hati dan memberitahu doktor yang bertanggungjawab sekiranya berlaku kelainan.
Secara umum, sindrom Muenke dapat dirawat dengan baik dan tidak mengakibatkan penurunan kualiti hidup secara kekal. Walau bagaimanapun, kecacatan dan kecacatan hampir selalu ada, yang kadang-kadang boleh menjadi beban emosi yang besar bagi mereka yang terjejas. Atas sebab ini, rawatan perubatan harus disokong oleh langkah-langkah terapi.
Kanak-kanak yang terjejas teruk dengan perbezaan perkembangan dinasihatkan lebih awal untuk menghadiri tadika khas dan kemudian sekolah khas. Sebaliknya, rujukan dibuat hari ini untuk dimasukkan ke sekolah arus perdana. Anak-anak tanpa kelainan lain boleh pergi ke sekolah normal dan lebih tinggi pula.
Terapi komprehensif diperlukan, terutama dalam hal sekatan pergerakan yang teruk. Ibu bapa boleh menyokong langkah-langkah ini dengan menyokong anak dalam kehidupan seharian. Biasanya langkah fisioterapeutik selanjutnya ditunjukkan.
Oleh kerana penyakit ini juga dapat menjadi beban yang besar bagi saudara-mara, ibu bapa dan rakan-rakan juga harus meminta bantuan terapi. Ahli terapi juga dapat menjalin hubungan dengan orang lain yang terjejas dan, jika perlu, merujuk ibu bapa ke kumpulan pertolongan diri.

.jpg)